
 搜索
搜索  数字报
数字报  深圳号
深圳号  投诉举报
投诉举报  深圳特区报首席记者 罗莉琼 通讯员 深儿医 文/图
深圳特区报首席记者 罗莉琼 通讯员 深儿医 文/图毫无征兆的情况下,突然手肿、腿肿,有时身上一团团的红斑,紧张时肚子疼到打滚呕吐……13岁的凡凡(化名),从小就被一种“怪病”困扰。在她6岁时,她的爸爸也因为这个病,突然出现喉头水肿而窒息身亡。妈妈带着她四处求医,但症状仍未得到改善。
近日,凡凡来深圳市儿童医院风湿免疫科就诊,“谜团”才被解开,原来是遗传性血管性水肿。这是一种极其罕见的遗传病,患病率在1/5万到1/10万,容易被误诊为过敏。经过治疗,凡凡再也没出现腹痛和红斑,复查指标也已趋于稳定。
父女俩经常莫名水肿,七年前父亲窒息身亡
从小,凡凡的身上总是有一团团的红色斑块。当地医院的医生怀疑是“食物过敏”。为此,她的食谱里再也没有出现过鸡蛋、牛奶、坚果、海鲜等等“发物”。可是,红色斑块仍然不时出现。10年前,凡凡的双手双脚、脸部、腹部甚至私密部位,开始反复出现不明原因的水肿。每次发作两三天后,肿胀会逐渐消失。前往多家医院就诊后,她被诊断为“过敏”“荨麻疹”,奇怪的是过敏原却始终找不到。
随着年龄增大,她的症状也越来越严重,每次发作还会伴有剧烈腹痛、呕吐。两年前,在一次严重的发作中,由于出现了肠梗阻,当地医院的医生不得已为她做了肠切除手术。可是,由于病因无法找出,凡凡的病情并没有得到有效控制。
“这十几年,我没有睡过一天安稳觉”,凡凡的母亲罗女士介绍,孩子父亲也有同样症状,7年前突发喉头水肿,伴有呼吸困难,最终没能抢救过来。去年底,凡凡术后再次出现了剧烈腹痛、手脚及面部水肿等症状,被送到深圳市儿童医院。
在详细了解孩子病史后,风湿免疫科专家初步考虑她患有罕见病遗传性血管性水肿。为了明确诊断,医生对凡凡及其家人进行了血清学检查和基因学检查。结果显示:患者的C4浓度0.06g/L↓,C1脂酶抑制物(C1-INH)浓度0.36g/L↑,均明显异常;基因学检查显示SERPING1基因突变,结合临床表现和检查结果确诊为遗传性血管性水肿(Ⅱ型)。
经过输注血浆以及缓激肽受体拮抗剂治疗,凡凡再也没出现腹痛和红斑,复查指标也已趋于稳定。最近又有好消息传来:遗传性血管性水肿的靶向治疗药物,将在今年3月1日纳入医保。届时凡凡的病情有望进一步好转,她们面临的经济压力也将大大减轻。
这种水肿可能致命,致死率最高达40%
“这病非常罕见,也容易误诊。不要说病人了,连很多基层医院的医生都没接触过”,深圳市儿童医院风湿免疫科主任医师夏宇介绍,遗传性血管性水肿是一种常染色体显性遗传疾病。患者由于体内缺乏C1酯酶抑制物或其功能存在缺陷,而导致四肢、颜面、生殖器、呼吸道和胃肠道黏膜等身体多个部位毫无征兆地发生急性水肿。该病的发病率在1/5万到1/10万之间。由于疾病罕见,目前公众和医生对该疾病知之甚少,患者常常遭遇误诊误治。数据显示,我国患者诊断率不足5%。患者从发病到明确诊断,平均需要13年之久。
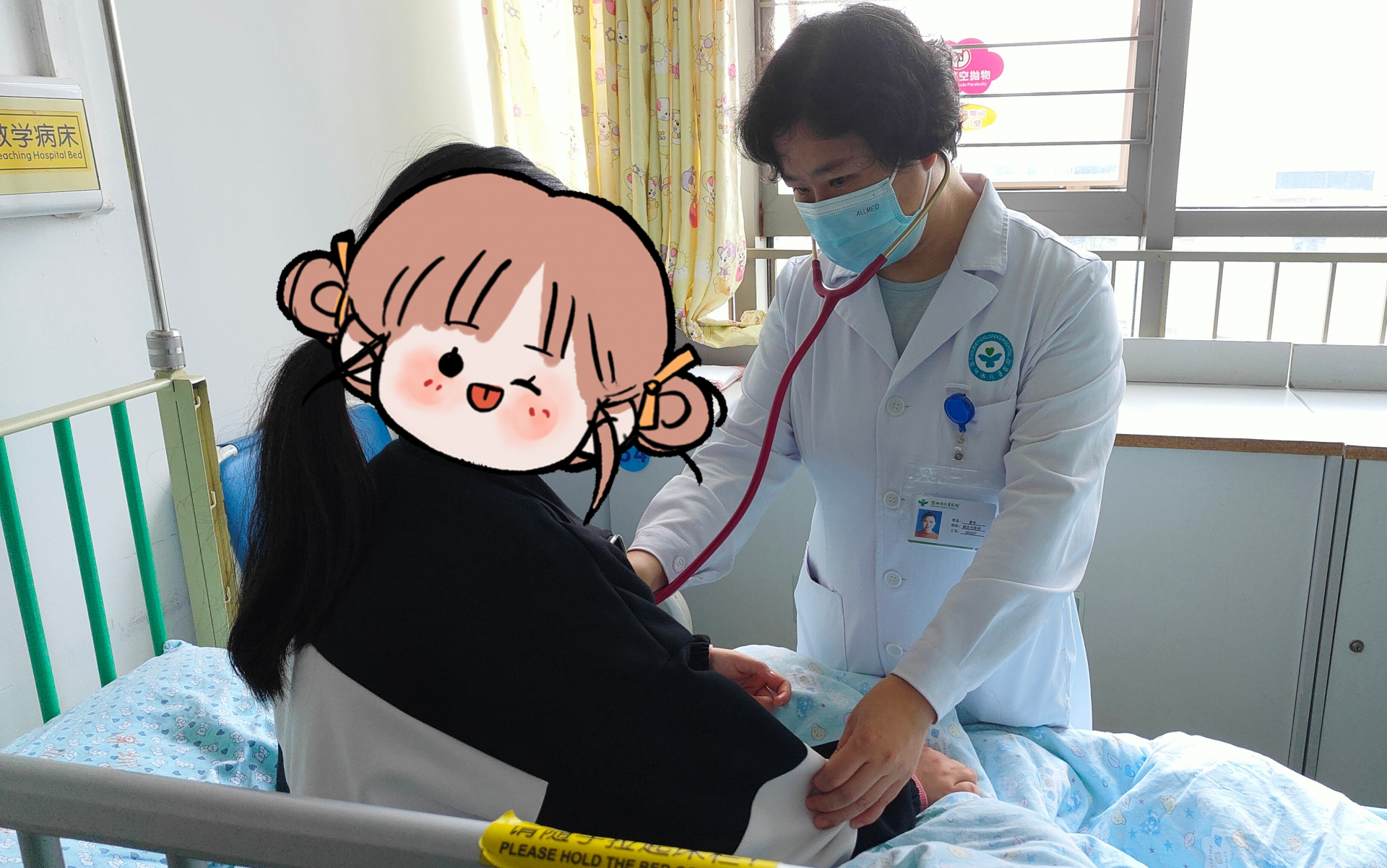
医生在为患儿凡凡复诊查体。
由于该病的发病不可预知,且发作时临床症状较为严重,甚至危及生命,患者应高度重视。其中,最为致命的是上呼吸道黏膜水肿,可迅速进展导致患者呼吸困难或窒息。据统计,我国有58.9%的遗传性血管性水肿患者发生过喉头水肿,其致死率最高可达40%。2018年,遗传性血管性水肿被收录我国《第一批罕见病目录》内。
【读特新闻+】
遗传性血管性水肿不是过敏,乱吃药无效且有风险
遗传性血管性水肿(英文简称“HAE”)发作时,水肿在全身多个部位都可能出现。该病发作的2—3天中,患者难以正常生活和社交。其中,胃肠道黏膜水肿发作时,会引起腹部剧烈疼痛且常伴有呕吐和/或腹泻。症状严重的患者,因剧烈的腹痛只能卧床休息,且通常需要72小时以上才能缓解。尤其值得警惕的是,上呼吸道黏膜水肿可迅速进展导致患者呼吸困难或窒息,危及生命。
受到水肿困扰时,有些“幸运”的HAE患者,吃了两天抗过敏药后身体见好,或者靠自己“硬扛”,水肿症状在3—5天后可能也缓解了。但事实上,这种自行缓解并非抗过敏药的“疗效”,抗过敏治疗对于遗传性血管性水肿是无效的,患者自行胡乱用药甚至可能会造成更严重的后果。
“长期有频繁水肿发作,又找不到原因的患者,不能仅考虑过敏,建议来完善相关检查”,夏宇医生介绍,临床诊断上,98%的患者都有C1-INH(C1酯酶抑制物)水平或者功能的异常,再结合临床症状,一般不难诊断。目前,深圳市儿童医院风湿免疫科可进行C1-INH相关检测。
(原标题《女孩经常莫名水肿 求医13年终在深圳找到病因》)
编辑 许家宜 审读 刘春生 二审 李怡天 三审 范京蓉
 报料
报料 


 读特热榜
读特热榜  IN视频
IN视频 




 鹏友圈
鹏友圈 




 iPhone客户端
iPhone客户端  安卓客户端
安卓客户端 
